Rối loạn chuyển hóa Lipid
- Bệnh Tay – Sachs (Bệnh rối loạn chuyển hóa ganglioside GM2 loại 1) (Hình 24.1)
Đặc điểm lâm sàng: bệnh thường biểu hiện vào tháng thứ 6 sau khi sinh. Trẻ đang khoẻ mạnh, tự nhiên yếu mệt, không đáp ứng với các kích thích thị giác, nhưng tăng đáp ứng với âm thanh. Một trong những dấu hiệu sớm nhất của bệnh là “Hoàng điểm đỏ anh đào”, do võng mạc vùng hoàng điểm mỏng và trong suốt cho phép nhìn thấy hắc mạc ở phía dưới. Các tế bào hạch xung quanh hoàng điểm bị nhiễm lipid GM2 tạo nên một vòng trắng quanh hoàng điểm. Vòng này thường nhạt màu dần và ngả sang màu hơi xám, trong khi đó đĩa thị trở nên bạc màu. Trẻ thường tử vong lúc 3 tuổi. Giải phẫu bệnh cho thấy não thường to, các tế bào hạch trương to chứa các thể trong bào tương. Các thể này cũng thấy trong các tế bào hạch ở võng mạc quanh hoàng điểm. Bệnh do thiếu men hexosaminidase A, làm cho lipid (GM2 ganliosid) tích luỹ bất thường trong tế bào, đặc biệt là tế bào thần kinh ở não. Hiện tượng tích luỹ lipid tăng dần sẽ gây tổn thương tiến triển cho tế bào. Hiện nay chưa có liệu pháp điều trị hiệu quả. Liệu pháp thay thế men hexosaminidase A cho trẻ bị thiếu men đã được đề xuất nhưng gặp phải khó khăn do men hexosaminidase A không qua được hàng rào máu não để đến được các tế bào não bị tổn thương.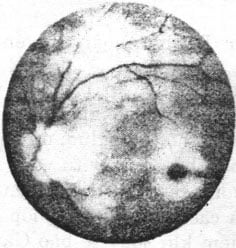
Hình 24.1. Bệnh Tay – Sachs “Hoàng điểm đỏ anh đào”
- Bệnh rối loạn chuyển hóa gangliosid GM2 loại 2 (Bệnh Sandhoff)
Di truyền: bệnh di truyền lặn liên kết nhiễm sắc thể thường. Bệnh thường gặp nhất ở người Do Thái. Bệnh cũng được phát hiện ở những chủng tộc khác. Xét nghiệm ước lượng hoạt tính men hexosaminidase A được dùng để phát hiện dị hợp tử và chẩn đoán trước khi sinh.
Triệu chứng ở mắt và não giống như bệnh Tay – Sachs nhưng có kèm tổn thương ở nội tạng do lắng đọng nhiều asilo GM2 và globosid. Xét nghiệm sinh hoá thấy thiếu cả men hexosaminidase A và B. Có thể chẩn đoán được trước khi sinh.
- Bệnh rối loạn chuyển hóa ganglioside GM2 loại 3
Trẻ bị bệnh thường biểu hiện triệu chứng lúc 2 – 5 tuổi. Trẻ bị mất điều hoà vận động, trì trệ phát triển tâm thần và thị lực kém. Tại mắt, đĩa thị và sắc tố võng nhạt màu. Trẻ thường tử vong lúc 5 – 15 tuổi. Chẩn đoán giải phẫu bệnh thấy lượng GM2 tăng quá mức. Nguyên nhân do thiếu một phần men hexosamidàse A. Có thể chẩn đoán người mang gen dị hợp tử và chẩn đoán trước khi sinh.
- Bệnh Gaucher (Bệnh rối loạn chuyển hóa glucosyl ceramid)
Bệnh này có ba thể: thể cấp, bán cấp, và mạn tính, cả ba thể đều đặc trưng bởi gan lách to, giảm tiểu cầu, giảm bạch cầu, có tế bào Gaucher trong hệ thống lưới nội mô. Thể cấp tính thường xảy ra ở trẻ còn bú, thể này gây tử vong nhanh. Thể bán cấp và thể mạn tính thường xảy ra ở thiếu niên. Hai thể này có mất tế bào thần kinh và các tế bào quanh lớp áo mạch máu ở não bị trường phồng. Hiếm khi thấy tế bào Gaucher ở não. Biểu hiện chính tại mắt là có thể xuất hiện các hạt (pingecula) màu nâu nhạt ở kết mạc và lác liệt trong thể cấp ở trẻ còn bú.
Sinh hoá: bệnh do thiếu men beta glucosidase. Men này có tác dụng tách glucosyl ceramid thành glucose và ceramid.
Di truyền: chưa rõ ràng, có thể có nhiều gen khác nhau tham gia gây ra ba thể bệnh khác nhau.
Nhưng có thể là di truyền lặn liên kêt nhiễm sắc thể thường. Thể cấp thường gặp ở người Do Thái. Có thể chẩn đoán người mang gen dị hợp tử và chẩn đoán trước khi sinh.
- Bệnh Fabry (Bệnh rối loạn chuyển hóa glycosphingolipid)
Đặc điểm lâm sàng: bệnh được Anderson và Fabry mô tả lần đầu tiên vào năm 1898. Bệnh biểu hiện ở nam với các triệu chứng đau nhức dữ dội ở các đầu ngón tay, đầu ngón chân, giãn mạch da, đặc biệt là vùng da từ rốn đến đầu gối. Có thể có giãn mạch ở miệng và kết mạc. Lắng đọng lipid ở thận gây protein niệu, lắng lipid ở hệ mạch gây nhồi máu cơ tim. Triệu chứng tại mắt bao gồm giãn các phình mạch ở tĩnh mạch kết mạc và võng mạc. Lắng đọng mỡ ở biểu mô giác mạc theo hình vòng xoắn điển hình giống trường hợp bị nhiễm chloroquin giác mạc. Thâm nhiễm giác mạc là dấu hiệu thường gặp nhất ở bệnh nhân nữ mang gen dị hợp tử. Mặc dù các dấu hiệu khác có thể có nhưng không rõ như ở người mang gen đồng hợp tử.
Sinh hoá: lipid lắng đọng là galactosyl glucosyl ceramid. Đây l à một trong những glycosphingolipid thần kinh. Bệnh do thiếu men alpha – galactosyl hydrolase.
Di truyền: bệnh di truyền lặn liên kết nhiễm sắc thể X. Có thể chẩn đoán bệnh trước khi sinh bằng chọc ối và xét nghiệm tế bào tìm lipid và men liên quan.
- Bệnh rối loạn chuyển hóa lipid sphingomyelin (Bệnh Niemann – Pick)
Bệnh thường gặp ở người Do Thái. Bệnh có ba thể chính là: the A, B, và c. Nguyên nhân do thiếu men sphingomyelinase, men này cần thiết cho quá trình chuyển hóa sphingomyelin, một thành phần của màng tế bào. Khi thiếu men này sphigomyelin sẽ bị lắng đọng bất thường trong tổ chức. Gen chi phối men sphingomyelin nằm ở nhiễm sắc thể 11.
Thể A là thể thần kinh cấp, gặp ở trẻ còn bú. Đây là thể thường gặp nhất. Bệnh thường bắt đầu vài tháng sau khi sinh. Trẻ khó bú sữa, gan lách to, mất dần khả năng vận động, trì trệ phát triển tâm thần. Đáy mắt có dấu hiệu hoàng điểm đỏ anh đào giống như bệnh Tay – Sachs do thâm nhiễm lipid quanh hoàng điểm. Trẻ thường tử vong khi được 3 đến 4 tuổi. Giải phẫu bệnh cho thấy có các tế bào bọt phân bố trong khắp hệ thống lưới nội mô, đặc biệt trong gan, lách, hạch và tủy xương. Trong các tổ chức nhãn cầu có các thể tương bào. Các thể này cũng thấy trong tế bào hạch, tế bào amacrin, tế bào Mũller, tế bào nón, tế bào gậy, tế bào biểu mô sắc tố, và trong hắc mạc, giác mạc, củng mạc.
Chẩn đoán dựa vào nuôi cấy nguyên bào sợi ở da hoặc bạch cầu và xét nghiệm tìm men sphingomyelinase. Xét nghiệm cho phép phát hiện những người mang gen dị hợp tử, và được dùng để chẩn đoán trước khi sinh.
Thể B là thể mạn tính. Triệu chứng chủ yếu là gan lách to, chậm phát triển, không hên quan đến hệ thần kinh trung ương. Thể c đặc trưng bởi các triệu chứng thần kinh trung ương mạn tính.
Di truyền: bệnh di truyền lặn liên kết nhiễm sắc thể thường.
- Bệnh lecithin mang tính gia đình
Bệnh được đặc trưng bởi thâm nhiễm giác mạc gần vùng rìa. Thiếu máu đẳng sắc và protein niệu. Xét nghiệm máu thấy lượng cholesterol ester giảm, nhưng cholesterol chưa ester hoá và các mỡ khác tăng.
Sinh hoá: bệnh do thiếu men cholesterol acyl transferase trong huyết tương. Men này cần thiết cho quá trình chuyển hóa các acid béo chưa bão hoà từ lecithin thành cholesterol.
Di truyền: bệnh di truyền lặn liên kết nhiễm sắc thể thường.
- Bệnh Wolman
Đây là bệnh nặng, xảy ra ở trẻ nhỏ, đặc trưng bởi gan lách to, tuyến thượng thận to và calci hoá. Lipid lắng đọng trong tế bào hạch võng mạc, củng mạc, giác mạc, và thể mi. Thừa lượng mỡ trung tính dự trữ, thừa cholesterol ester và triglycerid. Bệnh do thiếu men lipase.
Di truyền: bệnh di truyền lặn liên kết nhiễm sắc thể thường.
Rối loạn Lipoprotein huyết thanh
Các rối loạn chính trong nhóm liên quan đến triglycerid, cholesterol, và phospholipid. Các lipid này đều gắn với protein để tạo thành lipoprotein.
Các bệnh thiếu hụt lipoprotein.
Bệnh thiếu abetalipoprotein huyết (Hội chứng Bassen – Kornzweig)
Bệnh được đặc trưng bởi viêm võng mạc sắc tố, hồng cầu hình tế bào gai, mất điều hoà vận động, và phân có mỡ. Các dấu hiệu ở trẻ nhỏ bao gồm bụng chướng và phân có mỡ. Hồng cầu hình tế bào gai. Đáy mắt có hình ảnh điển hình của viêm võng mạc sắc tố, đĩa thị nhạt màu, mạch máu nhỏ, sắc tố lắng đọng dạng tế bào xương. Hiện tượng sắc tố lắng đọng ở hoàng điếm có thể xảy ra trước khi có các biến đổi ở võng mạc chu biên. Điện sinh lý cho thây điện thế giảm nặng. Sinh thiết hỗng tràng thấy các tế bào biểu mô dạng không bào và tế bào bọt điển hình. Chẩn đoán dựa vào xét nghiệm thấy nồng độ triglycerid và cholesterol huyết thanh thấp. Xét nghiệm sinh hoá miễn dịch không thấy betalipoprotein và apobetalipoprotein, hai protein tham gia vận chuyển lipid từ ruột vào máu.
Di truyền: bệnh di truyền lặn liên kết nhiễm sắc thể thường.