Bạch cầu cấp dòng tủy (AML) là bệnh lý ác tính theo dòng của tiền chất tủy xương dòng tủy, trong đó các tế bào kém biệt hóa tích tụ trong tủy xương và tuần hoàn.
Các triệu chứng xảy ra do sự vắng mặt của các tế bào trưởng thành bình thường do tủy xương sản xuất, bao gồm các bạch cầu hạt (nhạy cảm với nhiễm trùng) và tiểu cầu (nhạy cảm với xuất huyết). Hơn nữa, nếu số lượng lớn các nguyên bào tủy ác tính chưa trưởng thành lưu hành, chứng có thể xâm nhập tổ chức và hiếm khi gây rối loạn chức năng. Có các phân nhóm hình thái khác nhau (Bảng) có phần lớn các đặc điểm lâm sàng chồng chéo. Đáng chú ý là các bệnh nhân bạch cầu cấp thể tiền tủy bào (APL) (FAB M3) có xu hướng phát triển xuất huyết và đông máu nội mạch rải rác, đặc biệt trong khi cảm ứng hóa chất, do giải phóng các procoagulant từ các hạt trong bào tương.
Tỷ lệ mắc và bệnh nguyên
Ở Hoa Kỳ khoảng 13,780 trường hợp mắc năm 2012. Bạch cầu cấp dòng tủy chiếm khoảng 80% bạch cầu cấp ở người lớn. Bệnh nguyên chưa rõ ở đại đa số các trường hợp. Ba phơi nhiễm môi trường làm tăng nguy cơ: tiếp xúc benzen lâu dài, phơi nhiễm bức xạ và đã điều trị trước đó với tác nhân alkyl hóa (đặc biệt đồng thời xạ trị) và chất ức chế topoisomerase II (VD doxorubicin và etoposide). Bạch cầu kinh dòng tủy (CML), các hội chứng rối loạn sinh tủy và rối loạn tăng sinh tủy đều có thể tiển triển thành AML. Một số bất thường về gen nhất định có liên quan tới các biến thể hình thái đặc biệt: t (15;17) với APL, inv (16) với bệnh bạch cầu ái toan; các bất thường khác xảy ra ở nhiều thể. Nhiễm sắc thể 11q23 bất thường thường thấy ở bệnh bạch cầu tiến triển sau tiếp xúc với các chất ức chế topoisomerase II. Sự xóa bỏ nhiễm sắc thể 5 hoặc 7 được thấy ở bệnh bạch cầu sau xạ trị kết hợp hóa trị. Các bất thường về gen đặc biệt có ảnh hưởng mạnh mẽ đến kết quả điều trị. Biểu hiện của MDR1 (bơm kháng đa thuốc) thường gặp ở bệnh nhân cũ và ảnh hưởng xấu đến tiên lượng.
Bảng. HỆ THỐNG PHÂN LOẠI AML
Phân loại của tổ chức y tế thế giớia
AML có những bất thường di truyền tái phát
AML có t(8;21)(q22;q22);RUNX1-RUNX1T1b
AML có inv(16)(p13.1;1q22) hoặc t(16;16)(p13.1;q22);CBFB-MYH11b
Bạch cầu cấp thể tiền tủy bào có t(15;17)(q22;q12); PML-RARAb
AML có t(9;11)(p22;q23); MLLT3-MLL
AML có t(6;9)(p23;q34); DEK-NUP214
AML có inv(3)(q21q26.2) or t(3;3)(q21;q26.2); RPN1-EVI1
AML (nguyên mẫu tiểu cầu) có t(1;22)(p13;q13); RBM15-MKL1
Thực thể tạm thời: AML có biến đổi gen NPM1
Thực thể tạm thời: AML có biến đổi gen CEBPA
AML có những thay đổi liên quan đến rối loạn sinh tủy
U tủy liên quan đến điều trị
AML không xếp loại khác
AML biệt hóa tối thiểu
AML không trưởng thành
AML trưởng thành
Bạch cầu cấp dòng tủy-mono
Bạch cầu cấp dòng nguyên bạch cầu mono và bạch cầu mono
Bạch cầu cấp dòng hồng cầu
Bạch cầu cấp dòng mẫu tiểu cầu
Bạch cầu cấp dòng bạch cầu ái kiềm
Tăng sinh các tế bào tuỷ cấp có tăng sinh xơ tủy
Sarcom tủy
Tăng sinh dòng tủy liên quan đến hội chứng Down
Tạo tế bào tủy bất thường thoáng qua
Bệnh bạch cầu dòng tủy liên quan đến hội chứng Down
Tân sản tế bào tua non dạng tương bào
Bạch cầu cấp không rõ dòng
Bạch cầu cấp không biệt hóa
Bạch cầu cấp phối hợp phenotype t(9;22)(q34;q11,20); BCR-ABL11
Bạch cầu cấp phối hợp phenotype có t(v;11q23); MLL tái cấu trúc
Bạch cầu cấp phối hợp phenotype, B/tủy, NOS
Bạch cầu cấp phối hợp phenotype, T/tủy, NOS
Thực thể tạm: TB diệt tự nhiên (NK) Bệnh BC nguyên bào lympho/u lympho
Phân loại Pháp – Mỹ – Anh (FAB)c
MO: Bệnh bạch cầu biệt hóa tối thiểu
Ml: Bệnh bạch cầu nguyên tủy bào không trưởng thành
M2: Bệnh bạch cầu nguyên tủy bào trưởng thành
M3: Bệnh bạch cầu tiền tủy bào tăng bạch cầu hạt
M4: Bệnh bạch cầu dòng tủy – mono
M4Eo: Biến thể: Tăng bạch cầu ái toan tủy bất thường
M5: Bệnh bạch cầu dòng mono
M6: Bệnh bạch cầu dòng hồng cầu (Bệnh DiGuglielmo)
M7: Bệnh bạch cầu dòng mẫu tiểu cầu
aTheo SH Swerdlow và cộng sự (eds): Tổ chức Y tế Thế giới phân loại các khối u hệ tạo máu và mô lympho. Lyon, IARC Press, 2008.
bChẩn đoán là AML bất kể số lượng blast.
cTừ JM Bennett và cộng sự: Ann Intern Med 103:620, 1985.
Viết tắt: AML: bạch cầu cấp dòng tủy; BC: bạch cầu.
Đặc điểm lâm sàng và cận lâm sàng
Các triệu chứng đầu tiên của bạch cầu cấp thường biểu hiện khoảng < 3 tháng; hội chứng tiền lơ-xê-mi có thể gặp trong 25% số bệnh nhân bạch cầu cấp dòng tủy.
Triệu chứng thiếu máu, xanh xao, mệt mỏi, suy nhược, hồi hộp và khó thở khi gắng sức hay gặp nhất. Số lượng bạch cầu (WBC) có thể thấp, bình thường hoặc tăng rõ rệt; các tế bào blast có thể có hoặc không trong vòng tuần hoàn; với WBC >100 × 109 blast/l, có thể có tăng lắng đọng bạch cầu ở phổi và não. Nhiễm trùng mụn mủ nhỏ ở da hay gặp. Giảm tiểu cầu dẫn đến chảy máu tự phát, chảy máu cam, đốm xuất huyết, xuất huyết kết mạc, chảy máu lợi và bầm máu, đặc biệt khi số lượng tiểu cầu < 20,000/μL. Hay gặp chán ăn và gầy sút cân; có thể có sốt.
Nhiễm khuẩn và nhiễm nấm hay gặp; nguy cơ cao hơn khi số lượng bạch cầu trung tính < 5000/μL, tổn thương hàng rào da và niêm mạc làm trầm trọng thêm sự nhạy cảm; nhiễm trùng có thể bị che khuất trên lâm sàng do giảm bạch cầu trầm trọng và để nhận ra nhanh chóng đòi hỏi mức độ nghi ngờ lâm sàng cao.
Gan, lách to xảy ra ở khoảng một phần ba số bệnh nhân; viêm màng não do bệnh bạch cầu có thể có với triệu chứng đau đầu, buồn nôn, co giật, phù gai thị, liệt thần kinh sọ.
Các bất thường chuyển hóa bao gồm hạ natri máu, hạ kali máu, tăng lactate dehydrogenase (LDH) huyết thanh, tăng acid uric máu và nhiễm toan lactic (hiếm). Nếu số lượng tế bào blast rất cao trong máu, tăng kali máu giả và hạ đường huyết có thể xảy ra (kali được giải phóng và glucose bị tiêu thụ bởi các tế bào ung thư sau khi máu bị rút ra).
Điều trị bạch cầu cấp dòng tủy
Lượng tế bào bạch cầu tại thời điểm phát hiện có thể là 1011–1012 tế bào; khi số lượng bạch cầu giảm xuống dưới ~109, chúng không còn được phát hiện trong máu và tủy xương và bệnh nhân có vẻ thuyên giảm hoàn toàn (CR). Vì vậy điều trị tích cực phải tiếp tục qua thời điểm khi số lượng lớn các tế bào ban đầu giảm nếu bệnh bạch cầu được loại trừ. Giai đoạn hóa trị điển hình bao gồm điều trị cảm ứng thuyên giảm và hậu thuyên giảm, với việc điều trị kéo dài khoảng 1 năm. Hình chỉ ra phác đồ điều trị.
Chăm sóc hỗ trợ bằng truyền hồng cầu và tiểu cầu [từ người hiến có cytomegalovirus (CMV) huyết thanh âm tính, nếu bệnh nhân là ứng viên ghép tủy xương] là rất quan trọng, cũng như phòng chống, chẩn đoán và điều trị các bệnh nhiễm trùng tích cực. Các yếu tố kích thích dòng có ít hoặc không có lợi ích; một số khuyến cáo sử dụng ở những bệnh nhân lớn tuổi và những người có nhiễm trùng hoạt động. Giảm bạch cầu trung tính có sốt nên được điều trị bằng kháng sinh phổ rộng (VD ceftazidime 1g 8h/lần); nếu giảm bạch cầu trung tính có sốt kéo dài quá 7 ngày, có thể thêm amphotericin B.
Khoảng 60-80% bệnh nhân đạt được thuyên giảm ban đầu khi điều trị bằng cytarabine 100-200 (mg/m2)/ngày truyền liên tục trong 7 ngày và daunorubicin [45 (mg/m2)/ngày] hoặc idarubicin [12-13 (mg/m2)/ngày] trong 3 ngày. Thêm etoposide có thể cải thiện thời gian thuyên giảm hoàn toàn. Một nửa số bệnh nhân điều trị thuyên giảm hoàn toàn trong đợt hóa trị đầu tiên và 25% khác đạt được trong đợt 2. Khoảng 10-30% bệnh nhân có thời gian sống thêm 5 năm không bệnh và có thể chữa được.
Các bệnh nhân thuyên giảm hoàn toàn có nguy cơ tái phát thấp [các tế bào chứa t(8;21) hoặc inv(16)] nhận 3-4 đợt cytarabine. Những người có nguy cơ tái phát cao được cân nhắc cấy ghép tủy đồng loại.
Đáp ứng điều trị sau tái phát thường ngắn và tiên lượng những bệnh nhân tái phát thường kém. Trong APL, thêm trans-retinoic acid (tretinoin) và điều trị gây nên sự biệt hoá các tế bào bạch cầu và có thể cải thiện kết quả.
Arsenic trioxide cũng gây biệt hóa các tế bào APL.
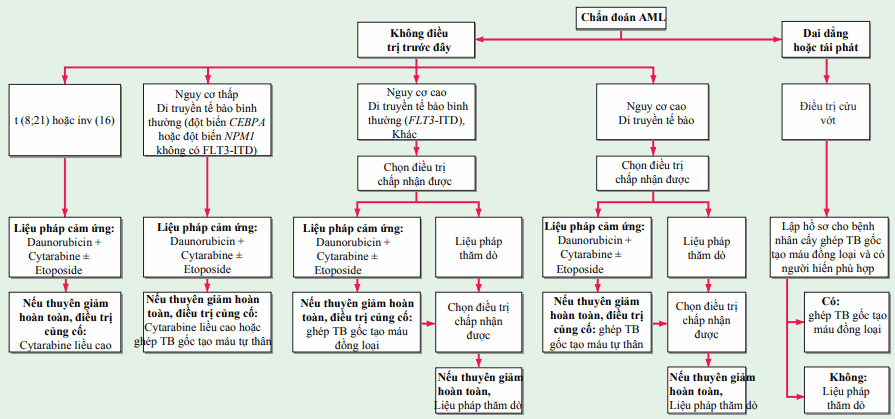
Hình. Lưu đồ điều trị cho chẩn đoán mới bạch cầu cấp dòng tủy. Với tất cả các thể AML trừ bạch cấu cấp thể tiền tủy bào (APL), liệu pháp chuẩn bao gồm truyền liên tục 7 ngày cytarabine (100–200 mg/m2/ngày) và một đợt 3 ngày daunorubicin (60–90 mg/m2/ngày) kèm theo có hoặc không 3 ngày etoposide (chỉ daunorubicin 60 mg/m2/ngày) hoặc liệu pháp mới dựa trên nguy cơ tái phát được dự đoán (tức là điều trị rủi ro phân tầng). Idarubicin (12–13 mg/m2/ngày) có thể thay thế daunorubicin (không hiển thị). Những bệnh nhân thuyên giảm hoàn toàn chuyển sang điều trị củng cố hậu thuyên giảm, gồm các đợt cytarabine liều cao liên tiếp, ghép tế bào gốc tạo máu (HSCT) tự thân, HSCT đồng loại, hoặc liệu pháp mới, dựa trên nguy cơ tái phát được dự đoán (tức là điều trị rủi ro phân tầng). Bệnh nhân có APL (xem sách về điều trị) thường dùng tretinoin đồng thời với hóa trị dựa trên anthracycline để thuyên giảm cảm ứng sau đó dùng arsenic trioxide kết hợp với hóa trị dựa trên anthracycline và có thể duy trì với tretinoin. Vai trò của cytarabine trong APL cảm ứng và kết hợp còn gây tranh cãi.
Ghép tủy xương từ người sinh đôi cùng trứng hoặc anh chị em có kháng nguyên bạch cầu người (HLA) giống hệt là cách điều trị hiệu quả cho bạch cầu cấp dòng tủy. Protocol điển hình dùng hóa trị liều cao ± xạ trị toàn thân để cắt bỏ tủy người nhận, sau đó truyền tủy người cho. Nguy cơ rất cao (trừ tủy từ người sinh đôi cùng trứng). Biến chứng gồm mô ghép chống vật chủ, viêm phổi kẽ, nhiễm trùng cơ hội (đặc biệt CMV). So sánh giữa cấy ghép và cytarabine liều cao như liệu pháp hậu thuyên giảm thấy không có lợi thế rõ ràng cho cách nào. Có đến 30% bệnh nhân bệnh bạch cầu dai dẳng giai đoạn cuối khác có thể chữa được nhờ cấy ghép; kết quả tốt hơn khi thực hiện cấy ghép trong khi thuyên giảm. Kết quả tốt nhất ở trẻ em và người trẻ tuổi.